Want to catch up with other articles from this series?
- The straight dope on cholesterol – Part I
- The straight dope on cholesterol – Part II
- The straight dope on cholesterol – Part III
- The straight dope on cholesterol – Part IV
- The straight dope on cholesterol – Part V
- The straight dope on cholesterol – Part VI
- The straight dope on cholesterol – Part VII
- The straight dope on cholesterol – Part VIII
- The straight dope on cholesterol – Part IX
Previously, in Part I, Part II and Part III of this series, we addressed these 5 concepts:
#1 — What is cholesterol?
#2 — What is the relationship between the cholesterol we eat and the cholesterol in our body?
#3 — Is cholesterol bad?
#4 — How does cholesterol move around our body?
#5 –How do we measure cholesterol?
Quick refresher on take-away points from previous posts, should you need it:
- Cholesterol is “just” another fancy organic molecule in our body but with an interesting distinction: we eat it, we make it, we store it, and we excrete it – all in different amounts.
- The pool of cholesterol in our body is essential for life. No cholesterol = no life.
- Cholesterol exists in 2 forms – unesterified or “free” (UC) and esterified (CE) – and the form determines if we can absorb it or not, or store it or not (among other things).
- Much of the cholesterol we eat is in the form of CE. It is not absorbed and is excreted by our gut (i.e., leaves our body in stool). The reason this occurs is that CE not only has to be de-esterified, but it competes for absorption with the vastly larger amounts of UC supplied by the biliary route.
- Re-absorption of the cholesterol we synthesize in our body (i.e., endogenous produced cholesterol) is the dominant source of the cholesterol in our body. That is, most of the cholesterol in our body was made by our body.
- The process of regulating cholesterol is very complex and multifaceted with multiple layers of control. I’ve only touched on the absorption side, but the synthesis side is also complex and highly regulated. You will discover that synthesis and absorption are very interrelated.
- Eating cholesterol has very little impact on the cholesterol levels in your body. This is a fact, not my opinion. Anyone who tells you different is, at best, ignorant of this topic. At worst, they are a deliberate charlatan. Years ago the Canadian Guidelines removed the limitation of dietary cholesterol. The rest of the world, especially the United States, needs to catch up. To see an important reference on this topic, please look here.
- Cholesterol and triglycerides are not soluble in plasma (i.e., they can’t dissolve in water) and are therefore said to be hydrophobic.
- To be carried anywhere in our body, say from your liver to your coronary artery, they need to be carried by a special protein-wrapped transport vessel called a lipoprotein.
- As these “ships” called lipoproteins leave the liver they undergo a process of maturation where they shed much of their triglyceride “cargo” in the form of free fatty acid, and doing so makes them smaller and richer in cholesterol.
- Special proteins, apoproteins, play an important role in moving lipoproteins around the body and facilitating their interactions with other cells. The most important of these are the apoB class, residing on VLDL, IDL, and LDL particles, and the apoA-I class, residing for the most part on the HDL particles.
- Cholesterol transport in plasma occurs in both directions, from the liver and small intestine towards the periphery and back to the liver and small intestine (the “gut”).
- The major function of the apoB-containing particles is to traffic energy (triglycerides) to muscles and phospholipids to all cells. Their cholesterol is trafficked back to the liver. The apoA-I containing particles traffic cholesterol to steroidogenic tissues, adipocytes (a storage organ for cholesterol ester) and ultimately back to the liver, gut, or steroidogenic tissue.
- All lipoproteins are part of the human lipid transportation system and work harmoniously together to efficiently traffic lipids. As you are probably starting to appreciate, the trafficking pattern is highly complex and the lipoproteins constantly exchange their core and surface lipids.
- The measurement of cholesterol has undergone a dramatic evolution over the past 70 years with technology at the heart of the advance.
- Currently, most people in the United States (and the world for that matter) undergo a “standard” lipid panel which only directly measures TC, TG, and HDL-C. LDL-C is measured or most often estimated.
- More advanced cholesterol measuring tests do exist to directly measure LDL-C (though none are standardized), along with the cholesterol content of other lipoproteins (e.g., VLDL, IDL) or lipoprotein subparticles.
- The most frequently used and guideline-recommended test that can count the number of LDL particles is either apolipoprotein B or LDL-P NMR which is part of the NMR LipoProfile. NMR can also measure the size of LDL and other lipoprotein particles, which is valuable for predicting insulin resistance in drug naïve patients (i.e., those patients not on cholesterol-lowering drugs), before changes are noted in glucose or insulin levels.
Concept #6 – How does cholesterol actually cause problems?
If you remember only one factoid from the previous three posts on this topic, remember this: If you were only “allowed” to know one metric to understand your risk of heart disease it would be the number of apoB particles (90-95% of which are LDLs) in your plasma. In practicality, there are two ways to do this:
- Directly measure (i.e., not estimate) the concentration of apoB in your plasma (several technologies and companies offer such an assay). Recall, there is one apoB molecule per particle;
- Directly measure the number of LDL particles in your plasma using NMR technology.
If this number is high, you are at risk of atherosclerosis. Everything else is secondary.
Does having lots of HDL particles help? Probably, especially if they are “functional” at carrying out reverse cholesterol transport, but it’s not clear if it matters when LDL particle count is low. In fact, while many drugs are known to increase the cholesterol content of HDL particles (i.e., HDL-C), not one to date has ever shown a benefit from doing so. Does having normal serum triglyceride levels matter? Probably, with “normal” being defined as < 70-100 mg/dL, though it’s not entirely clear this is an independent predictor of low risk. Does having a low level of LDL-C matter? Not if LDL-P (or apoB) are high, or better said, not when the two markers are discordant.
But why?
As with the previous topics in this series, this question is sufficiently complex to justify several textbooks – and it’s still not completely understood. My challenge, of course, is to convey the most important points in a fraction of that space and complexity.
Let’s focus, specifically, on the unfortunately-ubiquitous clinical condition of atherosclerosis – the accumulation of sterols and inflammatory cells within an artery wall which may (or may not) narrow the lumen of the artery.
Bonus concept: It’s important to keep in mind that this disease process is actually within the artery wall and it may or may not affect the arterial lumen, which is why angiograms can be normal in persons with advanced atherosclerosis. As plaque progresses it can encroach into the lumen leading to ischemia (i.e., lack of oxygen delivery to tissues) as the narrowing approaches 70-75%, or the plaque can rupture leading to a thrombosis: partial leading to ischemia or total leading to infarction (i.e., tissue death).
To be clear, statistically speaking, this condition (atherosclerotic induced ischemia or infarction) is the most common one that will result in the loss of your life. For most of us, atherosclerosis (most commonly within coronary arteries, but also carotid or cerebral arteries) is the leading cause of death, even ahead of all forms of cancer combined. Hence, it’s worth really understanding this problem.
In this week’s post I am going to focus exclusively on what I like to call the “jugular issue” – that is, I’m going to discuss the actual mechanism of atherosclerosis. I’m not going to discuss treatment (yet). We can’t get into treatment until we really understand the cause.
“It is in vain to speak of cures, or think of remedies, until such time as we have considered of the causes . . . cures must be imperfect, lame, and to no purpose, wherein the causes have not first been searched.”
—Robert Burton, The Anatomy of Melancholy, 1621
The sine qua non of atherosclerosis is the presence of sterols in arterial wall macrophages. Sterols are delivered to the arterial wall by the penetration of the endothelium by an apoB-containing lipoprotein, which transport the sterols. In other words, unless an apoB-containing lipoprotein particle violates the border created by an endothelium cell and the layer it protects, the media layer, there is no way atherogenesis occurs.
For now, let’s focus only on the most ubiquitous apoB-containing lipoprotein, the LDL particle. Yes, other lipoproteins also contain apoB (e.g., chylomicrons, remnant lipoproteins such as VLDL remnants, IDL and Lp(a)), but they are few in number relative to LDL particles. I will address them later.
The endothelium is the one-cell-thick-layer which lines the lumen (i.e., the “tube”) of a vessel, in this case, the artery. Since blood is in direct contact with this cell all the time, all lipoproteins – including LDL particles – come in constant contact with such cells.
So what drives an LDL particle to do something as sinister as to leave the waterway (i.e., the bloodstream) and “illegally” try to park at a dock (i.e., behind an endothelial cell)? Well, it is a gradient driven process which is why particle number is the key driving parameter.
As it turns out, this is probably a slightly less important question than the next one: what causes the LDL particle to stay there? In the parlance of our metaphor, not only do we want to know why the ship leaves the waterway to illegally park in the dock, but why does it stay parked there? This phenomenon is called “retention.”
Finally, if there was some way an LDL particle could violate the endothelium, AND be retained in the space behind the cell (away from the lumen on the side aptly called the sub-endothelial side) BUT not elicit an inflammatory (i.e., immune) response, would it matter?
I don’t know. But it seems that not long after an LDL particle gets into the sub-endothelial space and takes up “illegal” residence (i.e., binds to arterial wall proteoglycans), it is subject to oxidative forces and as one would expect an inflammatory response is initiated. The result is full blown mayhem. Immunologic gang warfare breaks out and cells called monocytes and macrophages and mast cells show up to investigate. When they arrive, and find the LDL particle, they do all they can to remove it. In some cases, when there are few LDL particles, the normal immune response is successful. But, it’s a numbers game. When LDL particle invasion becomes incessant, even if the immune cells can remove some of them, it becomes a losing proposition and the actual immune response to the initial problem becomes chronic and maladaptive and expands into the space between the endothelium and the media.
The multiple-sterol-laden macrophages or foam cells coalesce, recruit smooth muscle cells, induce microvascularization, and before you know it complex, inflamed plaque occurs. Microhemorrhages and microthrombus formations occur within the plaque. Ultimately the growing plaque invades the arterial lumen or ruptures into the lumen inducing luminal thrombosis. Direct luminal encroachment by plaque expansion or thrombus formation causes the lumen of the artery to narrow, which may or may not cause ischemia.
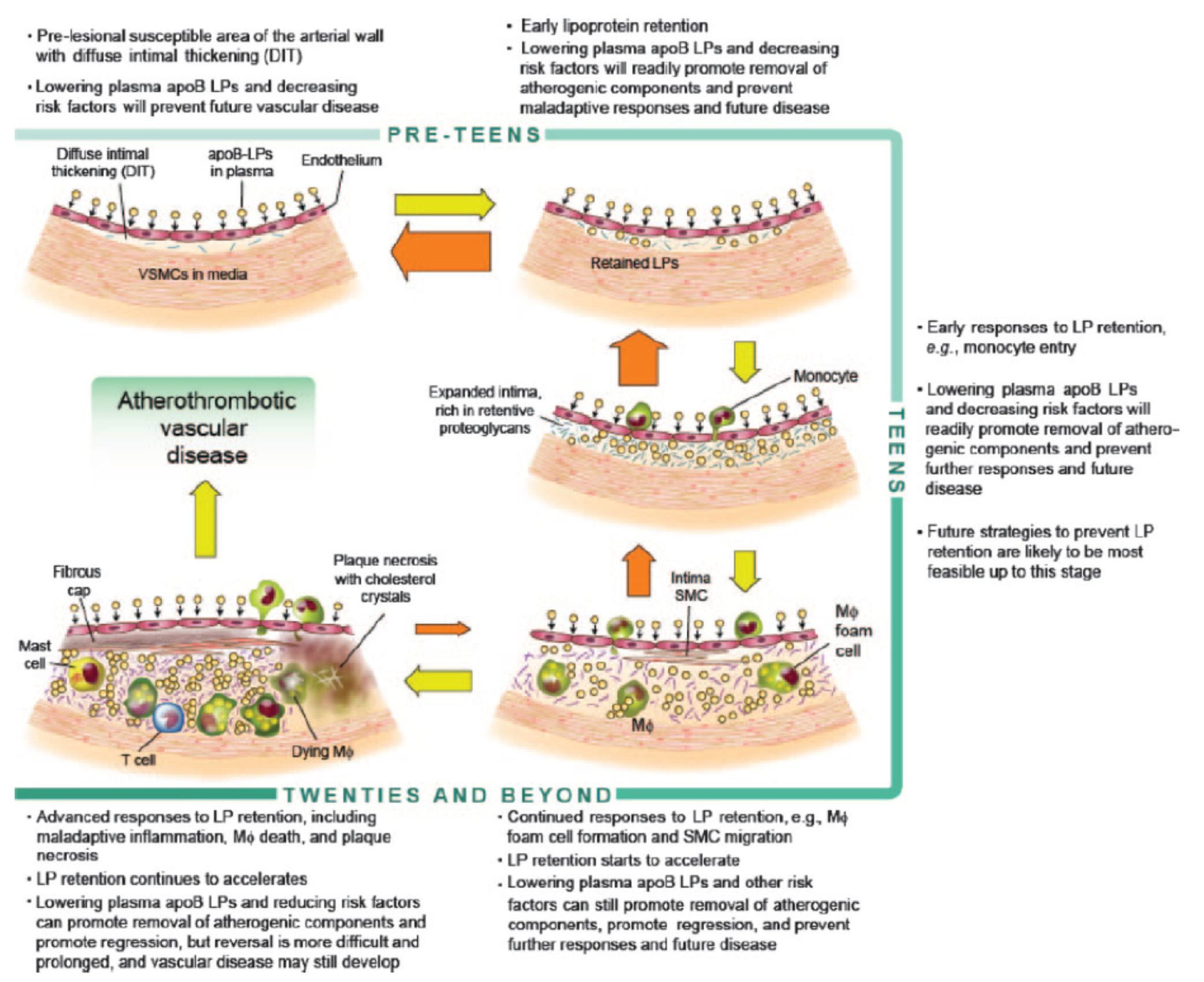
Before we go any further, take a look at the figure below from an excellent review article on this topic from the journal Circulation – Subendothelial Lipoprotein Retention as the Initiative Process in Atherosclerosis. This figure also discuss treatment strategies, but for now just focus on the pathogenesis (i.e., the cause of the problem).
In this figure you can see the progression:
- LDL particles (and a few other particles containing apoB) enter the sub-endothelium
- Some of these particles are retained, especially in areas where there is already a bit of extra space for them (called pre-lesion susceptible areas)
- “Early” immune cells initiate an inflammatory response which includes the direct interaction between the LDL particle and proteins called proteoglycans.
- The proteoglycans further shift the balance of LDL particle movement towards retention. Think of them as “cement” keeping the LDL particles and their cholesterol content in the sub-endothelial space.
- More and more apoB containing particles (i.e., LDL particles and the few other particles containing apoB) enter the sub-endothelial space and continue to be retained, due to the existing “room” being created by the immune response.
- As this process continues, an even more advanced form of immune response – mediated by cells called T-cells – leads to further retention and destruction of the artery wall.
- Eventually, not only does the lumen of the artery narrow, but a fibrous cap develops and plaques take form.
- It is most often these plaques that lead to myocardial infarction, or heart attacks, as they eventually dislodge and acutely obstruct blood flow to the portion of muscle supplied by the artery.
Another way to see this progression is shown in the figure below, which shows the histologic progression of atherosclerosis in the right coronary artery from human autopsy specimens. This figure is actually a bit confusing until you understand what you’re looking at. Each set of 3 pictures shows the same sample, but with a different kind of histological stain. Each row represents a different specimen.
- The column on the left uses a stain to highlight the distinction between the intimal and media layer of the artery call. The intima, recall, is the layer just below the endothelium and should not be as thick as shown here.
- The middle column uses a special stain to highlight the presence of lipids within the intimal layer.
- The right column uses yet a different stain to highlight the presence of macrophages in the intima and the media. Recall, macrophages are immune cells that play an important role of the inflammatory cascade leading to atherosclerosis.
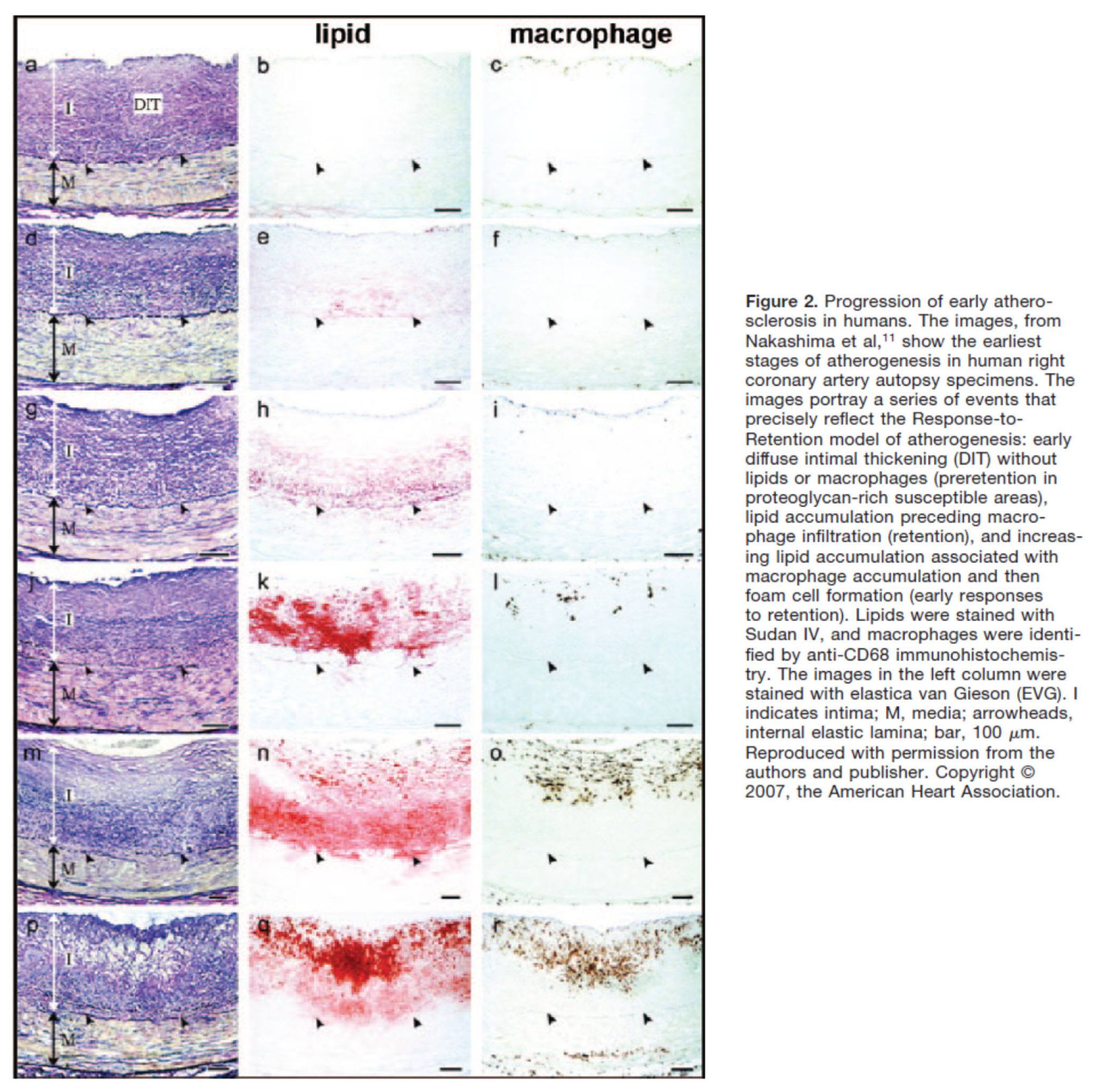
What is particularly compelling about this sequence of figures is that you can see the trigger of events from what is called diffuse intimal thickening (“DIT”), which exacerbates the retention of lipoproteins and their lipid cargo, only then to be followed by the arrival of immune cells, which ultimately lead the inflammatory changes responsible for atherosclerosis.
Why LDL-P matters most
You may be asking the chicken and egg question:
Which is the cause – the apoB containing LDL particle OR the immune cells that “overreact” to them and their lipid cargo?
You certainly wouldn’t be alone in asking this question, as many folks have debated this exact question for years. The reason, of course, it is so important to ask this question is captured by the Robert Burton quote, above. If you don’t know the cause, how can you treat the disease?
Empirically, we know that the most successful pharmacologic interventions demonstrated to reduce coronary artery disease are those that reduce LDL-P and thus delivery of sterols to the artery. (Of course, they do other things also, like lower LDL-C, and maybe even reduce inflammation.)
Perhaps more compelling is the “natural experiment” of people with genetic alterations leading to elevated or reduced LDL-P. Let’s consider an example of each:
- Cohen, et al. reported in the New England Journal of Medicine in 2006 on the cases of patients with mutations in an enzyme called proprotein convertase subtilisin type 9 or PCSK9 (try saying that 10 times fast). Normally, this proteolytic enzyme degrades LDL receptors on the liver. Patients with mutations (“nonsense mutations” to be technically correct, meaning the enzyme is somewhat less active) have less destruction of hepatic LDL receptors. Hence, they have more sustained expression of hepatic LDL receptors, improved LDL clearance from plasma and therefore fewer LDL particles. These patients have very low LDL-P and LDL-C concentrations (5-40 mg/dL) and very low incidence of heart disease. Note that a reduction in PCSK9 activity plays no role in reducing inflammation.
- Conversely, patients with familial hypercholesterolemia (known as FH) have the opposite problem. While there are several variants and causes of this disease, the common theme is having decreased clearance of apoB-containing particles, often but not always due to defective or absent LDL receptors, leading to the opposite problem from above. Namely, these patients have a higher number of circulating LDL particles, and as a result a much higher incidence of atherosclerosis.
So why does having an LDL-P of 2,000 nmol/L (95th percentile) increase the risk of atherosclerosis relative to, say, 1,000 nmol/L (20th percentile)? In the end, it’s a probabilistic game. The more particles – NOT cholesterol molecules within the particles and not the size of the LDL particles – you have, the more likely the chance a LDL-P is going to ding an endothelial cell, squeeze into the sub-endothelial space and begin the process of atherosclerosis.
What about the other apoB containing lipoproteins?
Beyond the LDL particle, other apoB-containing lipoproteins also play a role in the development of atherosclerosis, especially in an increasingly insulin resistant population like ours. In fact, there is some evidence that particle-for-particle Lp(a) is actually even more atherogenic than LDL (though we have far fewer of them). You’ll recall that Lp(a) is simply an LDL particle to which another protein called apoprotein(a) is attached, which is both a prothrombotic protein as well as a carrier of oxidized lipids – neither of which you want in a plaque. The apo(a) also retards clearance of Lp(a) thus enhancing LDL-P levels. It may foster greater penetration of the endothelium and/or greater retention within the sub-endothelial space and/or elicit an even greater immune response.
In summary
- The progression from a completely normal artery to an atherosclerotic one which may or may not be “clogged” follows a very clear path: an apoB containing particle gets past the endothelial layer into the sub-endothelial space, the particle and its cholesterol content is retained and oxidized, immune cells arrive, an initially-beneficial inflammatory response occurs that ultimately becomes maladaptive leading to complex plaque.
- While inflammation plays a key role in this process, it’s the penetration of the apoB particle, with its sterol passengers, of the endothelium and retention within the sub-endothelial space that drive the process.
- The most numerous apoB containing lipoprotein in this process is certainly the LDL particle, however Lp(a) (if present) and other apoB containing lipoproteins may play a role.
- If you want to stop atherosclerosis, you must lower the LDL particle number.
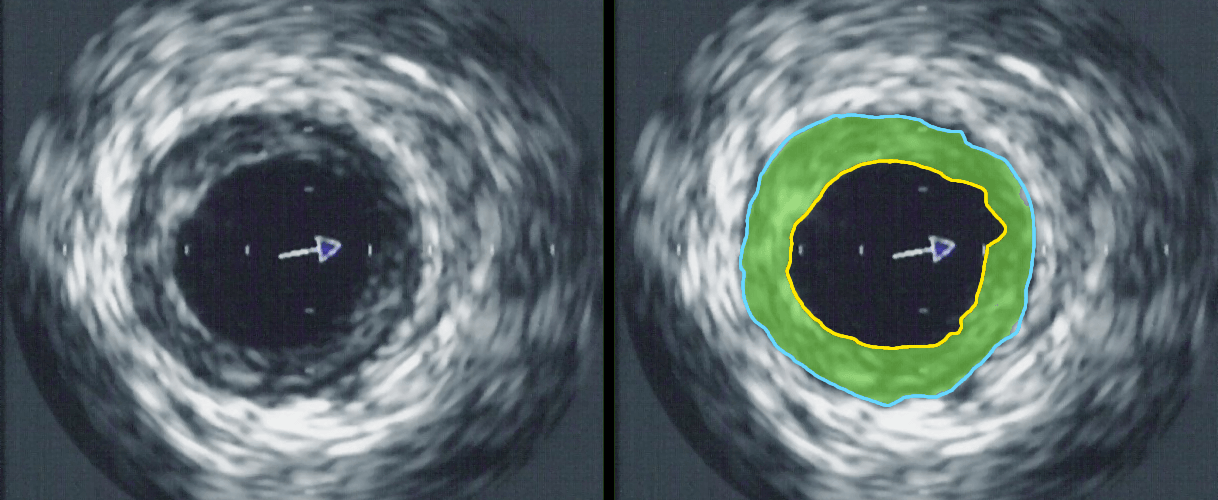
Intravascular ultrasound image of a coronary artery (left), with color coding on the right, delineating the lumen (yellow), external elastic membrane (blue) and the atherosclerotic plaque burden (green). The original uploader was Ksheka at English Wikipedia [CC BY-SA 2.5], via Wikimedia Commons

{kind=link}
Hi Dr [is the 11-th hour non stop, and i’m still reading your blog]
Pls correct the Robert Burton year he of “Anatomy of Melancholy, nice quote.”
He lived (8 February 1577 – 25 January 1640)
You have placed it in XIX cent.
cheers J.
Thanks, James. I think the year I have is the year the book was published from which I found it. Do you know the exact year?
You’re welcomed Doc. 1621 – was 1st publishing.
It was out of print from that time till ~1800., very much in demand.
No original copy survived, so all reprints are based on 1932.
As befitting of his time, R.B. was an erudite who studied from
physiology to astrology. He wrote a lot of latin poetry/ references in it.
“A-of-Mel” had many admirers from Samuel Johnson [who wrote “The Dictionary” Johnson claimed that he could finish it in 3-years. Academie Francaise used 40 scholars working for 40 yrs to finish theirs:) S-J said: “This is the proportion. 40 x 40 = 1600. As 3 goes to 1600, so is the proportion of an Englishman to a Frenchman”] to Keats, J.L.Borges etc.
Thank you!
Respose to Tim April 17, 2014
Cultures that do not eat heat processed unnatural polyunsaturated oils – heat processed refined sugars and non-fermented (soaked in water) grains and legumes –
have almost no heart problems –
The obvious conclusion being that diet and diet only is the most important thing in controlling risk –
Also – the LDL particle is composed of thousands of individual molecules – I assume some of these are glycated sugars and some polyunsaturated fats( if these things are in the diet) –
Opinion only – but in my mind – the glycated sugars with their hard-sharp molecular structures is what cuts into the artery wall and this starts a cascade of events – the polyunsaturated fats just being their act as the muck to help the repair –
So – the LDL Particle is not their to repair anything – toxic substances it is carrying create the problem and then the repair begins – being fixed or repaired with equally toxic stuff(oxidized polyunsatured oils)
The point being – no repair would have been needed if substances being carried by the LDL were not there
There are other factors involved as well – the general integrity of the bodies cells – calcium transport – correct immunne response
The number of LDL Particles therefore can only act as a guide – and simply implies that the more of them – the better chance the bad players attached to the LDL Particle will cause harm
If the LDL Particle is carring any virucs-bacteria-fungus-misshaped proteins: an increased foam cell response would occur
This is just how all this plays out in my mind – The LDL-Particle number between 15 year no cardiac event and those with an event is only on averaged out basis of 3000 people – 1300LDL-P to 1600LDL-P or not a great deal of difference
Since almost no one gets a test done for soft plaque build-up – a Doctor is left playing a guessing game – where a lousy LDL-P number is his best bet –
There’s one other theory worth looking at: processed food often contain hundreds of different egg sources(a commercial batch of cookies or bread or Miricle Whip and others containing egge sources from hundreds of different eggs) –
The possible problem being is that: certain egg proteins enter the blood stream and are certainly capable of attaching themselfs to an LDL-Particle – one egg would not be a problem – it’s the hundred’s of different egg sources that could cause a greater imunne response or some other metabolic havoc –
The person with the greatest imunne response to these hundred’s of DNA DIFFERENT egg proteins being at a higher risk –
It would seem a persons imunne response must play a role in soft plaque build-up –
jeff, could you provide references for this comment?
“Cultures that do not eat heat processed unnatural polyunsaturated oils – heat processed refined sugars and non-fermented (soaked in water) grains and legumes –
have almost no heart problems –
The obvious conclusion being that diet and diet only is the most important thing in controlling risk –”
scott becker – [email protected]
Hi Peter,
I am 48 years old and have hFH. I can’t tolerate statins, they make me depressed. 6 years ago I had a fast CT which revealed minimal plaque. I have been on a LCHF diet for about 11 years. From my understanding it appears that statins do not help in the case of FH since the cells are already depleted of cholesterol due to the lack of receptors. Statins would deplete the cells of vital cholesterol even further.
My interpretation of the overwhelming litterature is thus. A diet low in carbohydrate (less than 70 g per day), strictly limiting fructose, and PUFA, while high in saturated fat from nutrient dense animal foods, along with vegies is my best bet. Is this correct? I recently started eating more butter and my HDL skyrocketed. For the first time my ratios of TC to HDL are in the normal range given this increase in saturated fat. I also read recently, that when carbs are eaten, the best time of day is in the evening, since insulin produces a blunted response at that time. Thanks and keep up the good fight.
Maria, statins (and other lipid lowering meds) do help in FH, but they need to be the right drugs. Also, not clear what best diet is to minimize cholesterol synthesis. Varies by person, genes, etc.
Hi Peter,
Thanks so much for the education over the years. My families health is better for it and I don’t have to tell you what that’s worth.. I credit Taubes, you, Rosedale, Bernstein and a few others who can lay out this complex info in a way that makes sense to dopes like me so that we can apply the knowledge to improve our and our loved ones health.. Indeed, I’ve been able to “normalize” my boys T1D hbA1C – how awesome is that? 🙂 The state of medicine these days and the path that it’s got diabetics on (both t1 and t2) breaks my heart man.. But I digress…
I wanted to bring to your attention a fascinating paper by Vladimir M Subbotin
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3492120/
It corrects some common/persistent misperceptions of the coronary artery structure and offers an alternate hypothesis for the origin of atherosclerosis that answers a paradox or two that the current accepted model fails to explain.. It may have implications regarding this wonderful series you’ve written, maybe not.. I don’t think it’ll effect the focus I think you rightly put on particle count.. But it’s got important info in it, none the less.
Regardless, I think I understand you well enough to bet money that you’ll enjoy it if you have time. It’s not a long read.
Kind regards,
Kelly T
Ft. Collins CO
Look forward to checking it out.
Hey Peter,
I’m still in the middle of corresponding with the author, Mr. Subbotin, but it sounds to me like the established “gatekeepers” of funding and publications have tried to marginalize him. Hmm a clear thinking, rational, scientific mind dared to point out inaccuracies of an accepted premise, upon which the most profitable phara intervention known to man has since been built… Made me think of the arrows that you and Taubes take when you dare to ask rational, thoughtful questions that challenge the accepted dogma in the world of metabolism and nutrition..
When asked why he persists? “It is the meaning of my life.”
His answer reminded me of the “What’s your legacy?” T your wife gave ya.
Anyway. He sent me a link to a second paper he wrote on the subject.
https://www.tbiomed.com/content/4/1/41
It’s a real gem. Enjoy 😉
I read his paper in detail–with highlighter!–and I think he makes some interesting arguments. I guess my question is this, even if he is correct, does it change the role for LDL-P minimization?
It’s also possible that what occurs is a combo of both approaches (i.e., toward and from the lumen).
Whether or not this most interesting gentleman turns out to be correct, that he even *might* be correct shows that despite their high confidence Dayspring et al. have not by any stretch truly demonstrated that LDL-P is causative in CVD. This has always seemed apparent to me. A seemingly plausible mechanism + weak evidence ? proof of causation.
Even if LDL-P correlates better with CVD than LDL-C—and even if clinical trials show that pharmaceutically lowering LDL-P correlates better with improved outcomes than pharmaceutically lowering LDL-C—that in no way proves causation. Establishing basic biology takes an awful lot more than that. And without proven causation, and without clinical trials designed from the start to rigorously determine the risk/benefit of pharmaceutically lowering elevated LDL-P, how confident can any of us be that taking those drugs—which, to put it mildly, have non-trivial toxicity—is a wise move?
Hate to say it, but I can’t help comparing this state of affairs to Zoe Harcombe’s devastating demonstration of how little, if any, real science existed to support the radical dietary guidelines of the 1970’s. I’m not saying Dayspring et al. are necessarily wrong, or that no one should take statins, but how much we really know about these things is a question worth some serious consideration, IMO.
Not so, Bill. Nothing this fellow says disputes the idea that lipoprotein particles trafficking sterols into the subendothelial space causes atherosclerosis. The only point he is disputing, is the path they take to get there: do they diffuse from the lumen or do they get there from the circulation via capillaries.
In either case, fewer LDL-P is better, and this has been demonstrated by every study looking at CHD mortality vs. LDL-P.
I don’t know, Peter. You write, “Nothing this fellow says disputes the idea that lipoprotein particles trafficking sterols into the subendothelial space causes atherosclerosis.” But in my reading that’s exactly what he claims: “. . . logically, it must be concluded that high LDL levels are not ‘a major cause’ of coronary atherosclerosis. One of the major “assumptions” he pillories is that “a high level of LDL initiates and is the main cause of atherosclerosis.” Perhaps I recall incorrectly, but doesn’t Dr. Dayspring claim exactly what Subbotin denies—that LDL-P infiltration is the initiating and fundamental cause of atherosclerosis?
Subbotin does not deny that high LDL (I didn’t see him distinguishing between LDL-P and LDL-C) contributes to atherosclerosis, or that lowering LDL may modestly reduce the risk of getting CVD. But that’s not at all the same thing as accepting that LDL, in whatever form, is fundamentally causative of CVD.
Perhaps this seems like only a semantic difference, but I think Subbotin is right that it’s not, just as Gary Taubes was right that the mainstream energy balance model of obesity fundamentally and disastrously misconstrues the true causality. I love Subbotin’s quote from Stebhens:
“… differentiating between cause and non-causative factors is essential. Elimination of the latter only ameliorates or reduces the incidence whereas elimination of the former eradicates the disease. Swamps are not a cause of malaria. Draining swamps may reduce the incidence of malaria but it is eradication of the malarial parasites that eliminates the disease. Reduction in incidence rather than elimination of the disease precludes a causal relationship.”
Subbotin, in the second paper linked above, states without reservation, “We do not have a hint about the causative mechanisms.” Once we do, he thinks, perhaps there will be some truly effective strategies for preventing and treating CVD. But if we think we already know the underlying causal mechanism of course there won’t be much looking beyond it.
Other people, including Malcolm Kendrick and Carlos Monteiro, are also looking for alternate mechanisms of CVD in very serious ways. Jury still way out on the fundamental biology, to me at least.
In the meantime, maybe reducing LDL-P is a good idea for some of us, even with toxic drugs, in search of that possible modest reduction of risk. I’m not convinced, though your support this idea certainly makes me think twice. I’d like to see independent clinical trials testing the outcomes of mass screening of healthy populations for LDL-P, and treatment of those thereby deemed at risk. We’ve discussed this topic of screening before here. Past examples are not encouraging. As Dr. Gilbert Welch among others has shown, almost always huge numbers of healthy people are identified to be at risk and are exposed to the harms of intervention in order to, perhaps, help a much smaller number. The wisdom of this tradeoff is, to Welch and me and lots of others, far from clear. Maybe LDL-P screening will be different . . . anyway, interesting discussion, and I love Subbotin’s courage in challenging the orthodoxies of the scientific elites—kinda like you and Gary :-).
Dayspring (and every other lipidologist) argue that LDL-P enter the S-E space from the lumen (i.e., the shortest path). This author argues they get there by another path–namely through neovascularization through the entire wall thickness.
But they both agree that LDL particles end up the S-E space and that’s where the trouble takes place. What they disagree on is how they get there.
I’m not sure which one is correct and will always have sympathy for a contrarian view. But I don’t see how this alternative view alters the treatment, even if it is correct.
Thank you for the thoughtful discussion, gentlemen.
Two more papers that Mr. Subbotin has provided to me, both by William Stehbens. Do you have access to pubmed? I have these full pdf papers if not.. Anyone can ping me at my Kelly.trosper gmail account for the full papers.
Coronary heart disease, hypercholesterolemia, and atherosclerosis. I. False premises.
Coronary heart disease, hypercholesterolemia, and atherosclerosis. II. Misrepresented data.
Peter – “But I don’t see how this alternative view alters the treatment, even if it is correct.”
You’ve got a touching Tedx talk that demonstrates how important understanding the correct etiology of a condition/disease is when it comes to focusing on a target of treatment.. You went from a Dr. blaming the patient for being lazy and eating too much (calories-in/calories-out, exercise more – prevention/treatment for her diabesity), to understanding the etiology of obesity/diabetes (through your own personal journey) and eventually apologizing to her..
That took great courage and revealed your value for the truth.
You said you initially blamed her because you assumed the “pathological sequence of events was settled science.” And then, you went on to rattle off some very significant, important questions about diabetes and obesity that were only made possible by your efforts to step back and “question everything and challenge all assumptions…”
This may have profound implications to the target of treatment and current hypotheses, it might not. But the fact is, as long as current hypotheses are built on a faulty intellectual foundation (incorrect arterial morphology/design, mechanisms/progression of disease), science and treatment will never even think to focus on these other possible areas of treatment. We’re essentially throwing darts, hoping that something sticks.. (and sometimes they do.. never look a gift horse in the mouth, right?)
And you’re absolutely right. Particle count may be an important factor to control.. But if it is, it’s because we got lucky – not because we understand the true etiology of atherosclerosis…
Bill – “Subbotin, in the second paper linked above, states without reservation, “We do not have a hint about the causative mechanisms.” Once we do, he thinks, perhaps there will be some truly effective strategies for preventing and treating CVD.”
Yeah.
I see why he’s focused on educating the correct arterial morphology… That’s basically the lynch pin or the corner stone that current “pathological sequence of events and settled science” is built on.
If the correct morphology is agreed upon, taught/understood, better questions would necessarily be asked. Current hypotheses would no longer make sense, better ones would come along to explain the paradigms that aren’t even visible to these Docs and scientists who are currently pushing research/treatment based on an incorrect model down the wrong road…
Bill – ” I love Subbotin’s courage in challenging the orthodoxies of the scientific elites—kinda like you and Gary :-).”
Exactly. That’s why I got goose bumps when I stumbled upon his first paper..
I think of how Taubes’, in his book “Good Calories, Bad Calories”, documents the initial inertia built up by the (faulty and fraudulent) lipid hypothesis… How quickly it was backed by the entire weight of the established political, medical, nutritional and scientific communities..
That inertia, as you know, continues to this day and at times seems unrelenting.. It’s has pulled and is continuing to pull most research, science and medicine down a fruitless, harmful path.. not to mention the damage it’s causing the growing population of metabolically deranged people in this world..
Is it deja vu, all over again, with regard to Atherosclerosis?
Peter, this guy is fascinating to speak to.. I think the implications here can’t be overstated..
Would you consider emailing him and just having a conversation?? Wherever it might take you?
I have his contact info.
Quid enim iustificabit vitam tuam?
Of course, always happy to discuss.
Kelly,
Thanks for the studies you’ve listed!
Currently fighting to apply a keto-adapted paleo autoimmune diet for a 7 year old nephew/godson, and am mounting evidence that fats are good (grew up on a farm in NH and ate plenty fat, eggs, cream, etc and at 46 my health is at it’s prime, because of the evolutionary oriented mindset I’ve always had).
If you don’t mind, could you please email those study pdf’s to me? I will email you with “William E. Stehbens studies” in the subject line shortly.
Thanks Peter for a great lead of thought and science here!
Cheers,
Chris
Dear Peter,
First, thank you for this simply outstanding series. I haven’t found such a detailed cause-effect explanation anywhere else.
Like most other folks here, I have a question which I am currently unable to reconcile, but highly relevant to me, as my father died from a heart attack at 45, and his brother at 38; as I am 31, the clock is ticking.
I have been following a roughly Paleo / bulletproof diet for approximately two years, and each blood test has revealed a pattern of “relatively” low LDL-P_NMR (the most recent test show 1,804 nmol/L), but still high ApoB (102 mg/dL). [For reference, Large HDL-P: 4,386 nmol/L, and ‘high’ and ‘low’ are based upon the ranges provided by WellnessFX]. I wasn’t easily able to find the information to do a conversion, but if the ratio of LDL partcles to ApoB is close to ~1, I wouldn’t have expected to find this discrepancy.
You can find my full bloodwork here (though this link will expire after a few days):
https://awesomescreenshot.com/02f4rb0k3c
I found a recent article (https://www.lipidjournal.com/article/S1933-2874(14)00404-8/abstract?cc=y) suggesting that ApoB > LDL-P may indicate a higher-than-average prevalence of other non-LDL ApoB containing lipoproteins: however, my Lp(a) is low (21 mg/dL), sLDL (178), Medium LDL (393), TG (68), total Cholesterol 247, and phenotype A. I do not believe this makes up the discrepancy, though perhaps I am mistaken. The paper goes on to suggest to note that ApoB>LDL-P is most often associated with increased Lp(a) mass, but for me here that is not the case.
The closest analgous situation I could find was located at https://www.quantifiedbob.com/2014/09/bulletproof-diet-intermittent-fasting-1-5-year-results/, where the increases in lipoprotein markers were caused by other issues, such as chronic infection and thyroid issues (as an aside, I have suspect I may have some thyroid issues for some time; though my TSH looks acceptable, I suffer from dry skin and for the past six months have been shedding hair from my head, with no history of genetic baldness). I am also carrier for alpha-1 anti-trypsan deficiency (SZ); I don’t know if that plays any role; recent work suggest that it may do something (https://www.ncbi.nlm.nih.gov/pubmed/6333180), but most work has only been done on those with the full-blown version, not just carriers.
I’d love to get your thoughts on this. My PCP and cardiologist just want me on a low-dose statin. Would you recommend futher stool and/or saliva tests for investigation of thyroid and/or chronic infection? Is there someone in the Chicago area you would recommend seeing?
Apologies for the long post. Even if you don’t have time to answer, thanks again for all the work you do.
-Brendan
Brendan you MUST see a bulge bracket lipidologist. Someone needs to figure out your Lp(a)-P (not just cholesterol content), apoE, etc.
Hi Peter,
Sorry, what is a ‘bulge bracket lipidologist’? You mean someone who is well-known and respected? Can you give me names? The two folks I’m scheduling now are:
https://www.feinberg.northwestern.edu/faculty-profiles/az/profile.html?xid=22239 (he’d like me on a low-dose statin)
and
https://www.feinberg.northwestern.edu/faculty-profiles/az/profile.html?xid=17759 (haven’t spoken with him yet)
Thanks,
Brendan
Renowned lipidologist Dr. Tara Dall does phone consults on advanced testing and interpretation. She can be contacted through her website taradall.com. Best wishes, maryann
Yes, Dr. D is great.
Thanks very much to you both – I will schedule a call ASAP.
Hi Peter, Love your blog and the heavy science!!
I have been testing my lipids using NMR for years and my numbers are extremely high and not improving:
~2600 for LDL-p total, ~1800 LDL-P small. My wife (internal medicine) is fighting with me to take statins; as a clinical pharmacologist, I know the harm especially with no benefit. My TG is high (200-300), but not diabetic.
I feel I am a healthy 60 y old, carotid artery are clean, thinking about coronary cat scan? waiting for APOe test; but still at loss what path to take or how to interpret the results.
much appreciated…
keep the good research.
Hello Peter,
You have been a wealth of knowledge and I’ve read many of your blogs and watched you tube videos of your talks. I’ve looked for information regarding other markers in the Lipid Panel, such as Uric Acid and ALT and could find any associate blog posts. The reason I ask is because I just received my blood work back and both were flagged out of range high. I’ve read that Uric Acid has a tendency to go high, but after the adaptation it should go back down to normal levels, I’m at 9.7. I’m not sure about the ALT. Its a marker for liver damage as you know, but I haven’t been drinking and was only a social drinker before. My ALT was 48. I do carry extra weight. I’m about 30% BF right now. Working to bring that back down to 10% where I was just 4 years prior.
I’ve been eating high fat/low carb for 3 weeks now. Just adjusted my carbs from 10% to 5%.
Do you have any comments for people who may see abnormal Uric Acid and ALT numbers when beginning a ketogenic diet?
In addition, my LDL went above high for the first time, 106. But I already plan to ask for the NMR when I go to see my doctor for my annual physical.
Thanks for all you do!
-Nick
Maybe I missed this somewhere thoughout this series but what is the mechanism of high sugar/carb intake that drives elevation of LDL-P? I understand as we become more insulin resistant this number goes up but how does high blood sugar do this?
Great Info…
So it seems that if you are measured (by typical blood panel) to have low or normal LDL-C, you may still be at risk if your LDL-P is high. But, is the reverse scenario ever possible: where you are measured to have high LDL-C (by typical blood panel) but after an NMR the findings are that your LDL-P is low or within limits. Is this scenario even possible and would this mean you are, in fact, low risk for atherosclerosis?
I don’t see an answer to what causes the LDL to enter the sub-endothelial space in the first place. Or in your words “So what drives an LDL particle to do something as sinister as to leave the waterway (i.e., the bloodstream) and “illegally” try to park at a dock (i.e., behind an endothelial cell)?”
It looks to me like you’re saying that LDL itself is THE cause and therefore LDL, intrinsically, in and of itself is harmful to the arterial walls. But, even if that is what you’re saying, I see no explanation of a mechanism that initially causes, leads to or allows the LDL invasion. Do we know of a mechanism or is it all association at this point?
Also, if LDL itself is inherently harmful to arterial walls, why does the body make it and then use it in an environment where it will cause harm?
Please elucidate. Much appreciated!